A maioria dos casos de anemia é causada por deficiência de ferro na dieta. Mas algumas formas de anemia são hereditárias, isto é, anemia genética. A anemia é uma condição de saúde comum que às vezes pode incluir sintomas relativamente preocupantes. Sentir-se cansado, com baixa energia, memória fraca ou com falta de ar com dificuldade para exercícios são sintomas comuns associados à anemia.
Se você está se perguntando se tem ou não uma forma de anemia relacionada à sua genética, conversar com um membro da família pode ser um bom começo. Se um parente próximo tem um tipo genético de anemia, você também tem mais probabilidade de herdar anemia.
Mas para a maioria das pessoas, a anemia está relacionada a deficiências de nutrientes — não à genética.
A anemia é genética?
A maioria das formas de anemia não é devida à sua genética, mas vários tipos diferentes de anemia podem ser herdados. Na maioria dos casos, a anemia relacionada à sua genética é diagnosticada no nascimento ou identificada em uma idade jovem devido ao impacto relativamente grave que pode ter em seu corpo.
Que tipo de anemia é hereditária?
Vários tipos de anemia podem ser herdados. O plano de tratamento e a gravidade dos sintomas para essas doenças variam. Geralmente, os sintomas de anemia hereditária aparecem na infância.
Anemia falciforme
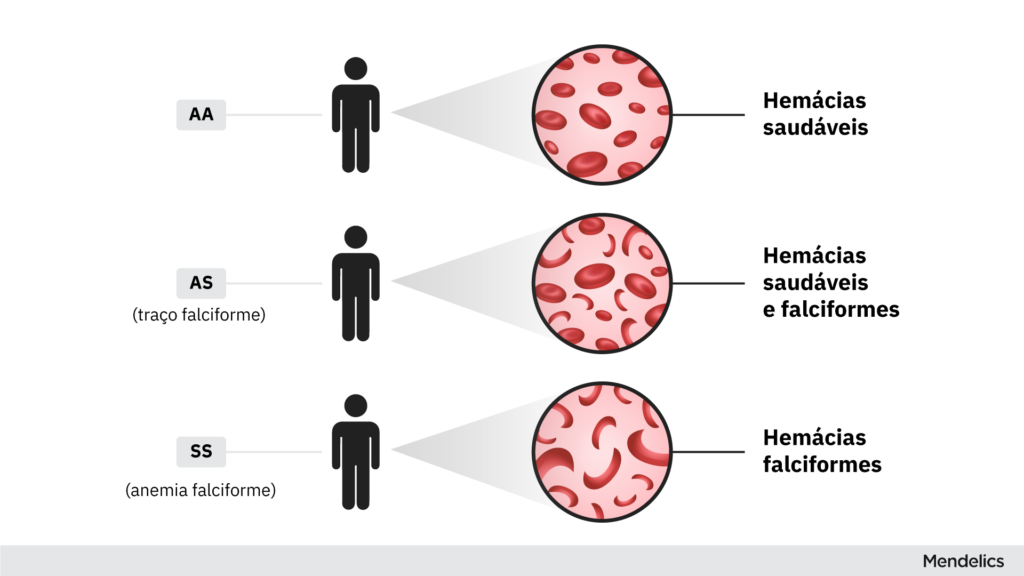
Frequentemente afetando afro-americanos, a anemia falciforme é o distúrbio sanguíneo hereditário mais comumente. A anemia falciforme afeta uma importante proteína do sangue chamada hemoglobina.
Como resultado, suas células sanguíneas assumem um formato crescente em vez do formato usual de disco. O resultado final dessa diferença estrutural é que seus glóbulos vermelhos se aglomeram mais facilmente.
Isso pode causar problemas sérios como derrame ou problemas oculares, e as células sanguíneas são destruídas muito mais rapidamente do que as células em formato de disco. É uma doença grave.
Talassemia
Semelhante à anemia falciforme, a talassemia se refere a um grupo de condições que afetam a proteína hemoglobina de maneiras específicas.
Certos subtipos de talassemia, como alfa e beta, são diagnosticados com base em como a proteína hemoglobina é alterada. Esse distúrbio geralmente é diagnosticado na primeira infância devido a sinais como crescimento lento ou ossos quebradiços.
Alguns tipos de talassemia são piores do que outros e podem produzir sintomas mais graves ou exigir tratamentos mais agressivos.
Esferocitose hereditária
A esferocitose hereditária ocorre quando seus glóbulos vermelhos perdem o formato de disco, mas o motivo pelo qual perdem o formato é diferente do que na talassemia e na anemia falciforme.

Enquanto a anemia falciforme e a talassemia resultam de alterações na hemoglobina, a esferocitose resulta de alterações em várias outras proteínas dos glóbulos vermelhos, como a espectrina.
Além da esferocitose, ocorrem outros tipos de doenças na hemácias causadas por alterações na sua forma. Entre elas, eliptocitose, piro-poiquilocitose, entre outras. O diagnóstico requer a demonstração do aumento da fragilidade osmótica dos eritrócitos e teste de antiglobulina direto negativo. Raramente, os pacientes com < 45 anos de idade com doença.
Deficiência de glicose-6-fosfato desidrogenase
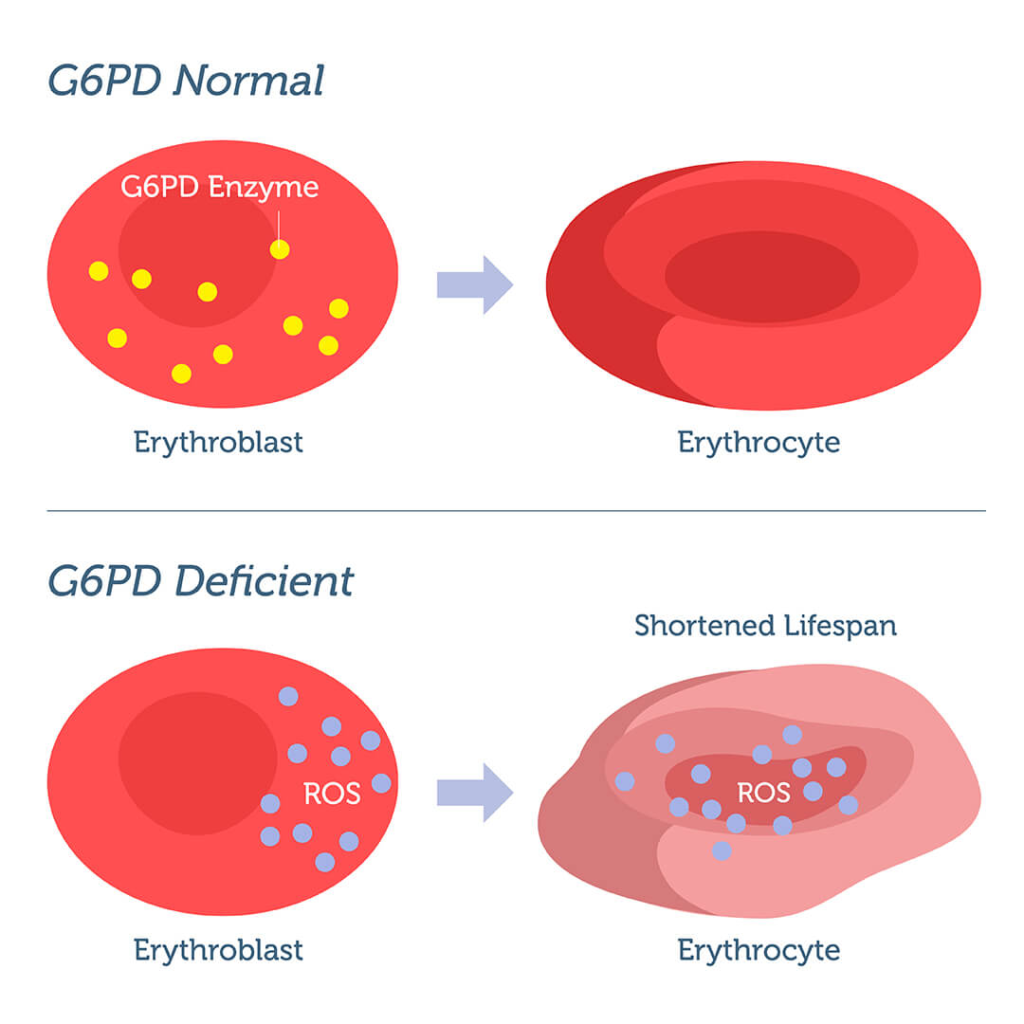
Também abreviado como deficiência de G6PD, esse distúrbio resulta da ausência de uma enzima que desempenha um papel importante na proteção dos glóbulos vermelhos contra danos. Sem essa enzima, os glóbulos vermelhos se tornam suscetíveis a danos e destruição excessiva.
A deficiência de G6PD é uma característica genética polimórfica que afeta mais de 500 milhões de pessoas no mundo todo. A maioria das pessoas com deficiência de G6PD é assintomática, a menos e até que sejam expostas a um agente exógeno, como uma refeição de favas, que desencadeia anemia hemolítica aguda. Essa crise hemolítica pode ser grave e pode exigir transfusão de sangue urgente. Manifestações menos comuns da deficiência de G6PD incluem anemia hemolítica não esferocítica crônica e icterícia neonatal.
Deficiência de Piruvato Quinase
A deficiência de piruvato quinase é uma doença rara causada por variantes autossômicas recessivas no gene PKLR, que codifica uma piruvato quinase que catalisa a transfosforilação do fosfoenolpiruvato em piruvato e adenosina trifosfato, a etapa limitante da taxa de glicólise. Embora a anemia hemolítica crônica seja a manifestação típica da deficiência de piruvato quinase, a apresentação clínica e as complicações variam consideravelmente de acordo com a idade.
O diagnóstico deve ser suspeitado na presença de sinais e sintomas clínicos e marcadores laboratoriais de anemia hemolítica crônica, incluindo esplenomegalia, icterícia, cálculos biliares, aumento de reticulócitos, hiperbilirrubinemia indireta e hiperferritinemia leve. A apresentação clínica pode ser diferente por faixa etária. Como a PKD é uma doença autossômica recessiva, o histórico familiar geralmente não é revelador, com exceção de abortos espontâneos e irmãos afetados.
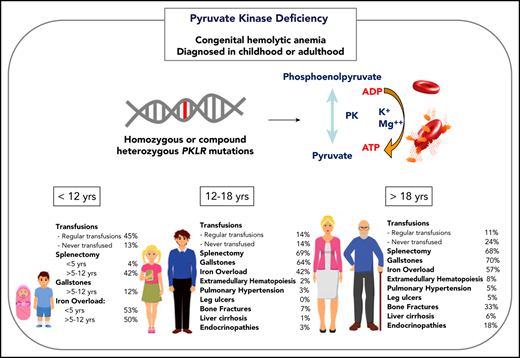
A apresentação clínica da PKD é variável e varia de complicações no útero a hiperbilirrubinemia indireta ou reticulocitose incidentalmente observadas associadas à hemólise totalmente compensada sem anemia, a anemia sintomática que leva a transfusões de sangue regulares. Os pacientes geralmente se apresentam no primeiro mês de vida; no entanto, os diagnósticos na idade adulta não são incomuns, particularmente em pacientes com hemólise compensada ou anemia leve ou que foram diagnosticados erroneamente com outro distúrbio hemolítico.
Insuficiência congênita da medula óssea
Como o nome sugere, a insuficiência da medula óssea ocorre quando a medula óssea não produz células sanguíneas suficientes. A medula óssea é onde a maioria das células sanguíneas é produzida. Existem diferentes tipos de insuficiência da medula óssea, como a Dimond-Black-Fan e Fanconi.
A anemia de Fanconi é a causa mais comum de insuficiência da medula óssea hereditária devido a um raro distúrbio genético autossômico recessivo envolvendo todas as 3 linhagens de células sanguíneas nas quais mutações homozigotas ou heterozigotas resultam em alelos patogênicos, incluindo mutações pontuais, duplicações, defeitos de splicing e deleções. Essas mutações genéticas dos genes da anemia de Fanconi causam um acúmulo de danos cromossômicos devido à incapacidade da célula de conduzir reparos. Essas mutações genéticas na via da anemia de Fanconi levam a células que não conseguem reparar adequadamente os danos ao DNA, resultando em instabilidade genômica, pancitopenia subsequente e maior suscetibilidade a agentes citotóxicos, radiação ultravioleta, deformação espontânea e predisposição a malignidades. Além disso, a anemia de Fanconi afeta quase todos os órgãos do corpo.
A anemia de Fanconi também é considerada uma forma hereditária de anemia aplástica. O distúrbio também pode predispor os pacientes ao desenvolvimento de tumores hematológicos e sólidos. Um diagnóstico de anemia de Fanconi é normalmente confirmado com um teste de fragilidade cromossômica, que continua sendo o padrão ouro. O tratamento da anemia de Fanconi inclui principalmente terapia de suporte, transplante de células-tronco hematopoiéticas e tratamento com andrógenos. Esta atividade para profissionais de saúde visa aumentar a competência dos alunos na seleção de testes diagnósticos apropriados, no tratamento da anemia de Fanconi e no fomento do trabalho em equipe interprofissional eficaz para melhorar os resultados.
A anemia de Diamond-Blackfan (DBA) é uma aplasia eritroide hereditária. Isto é, devido alterações genéticas, esses indivíduos não produzem globulos vermelhos. Esses pacientes possuem uma “eritroblastopenia” e representa a primeira ribossomopatia humana descrita. Um defeito na maturação do RNA ribossômico (rRNA) gera um estresse nucleolar resultando na parada do ciclo celular e apoptose de células eritroides, levando à ausência de produção de hemacias. O transplante alogênico de células-tronco hematopoiéticas ainda é o único tratamento curativo para DBA, enquanto glicocorticoides, transfusão de hemácias e terapia de quelação de ferro representam abordagens terapêuticas conservadoras
Anemia ferropriva refratária ao ferro
Há um caso específico de anemia ferropriva causada pela genética. Isso é conhecido como anemia ferropriva refratária ao ferro, que resulta em uma escassez de ferro no seu corpo. Para esse tipo de anemia, tomar um suplemento de ferro ou comer alimentos ricos em ferro não aumentará seus níveis de ferro.
A anemia ferropriva nem sempre resulta em sintomas perceptíveis. Você pode ser diagnosticado com base puramente em um resultado laboratorial mostrando baixo teor de ferro e alterações em seus glóbulos vermelhos.
Se ocorrerem sintomas, eles podem ser numerosos e vagos, mas há alguns sinais e sintomas reveladores que geralmente incluem:
- fadiga
- pele pálida
- fraqueza generalizada
- falta de ar
- tontura
- sensação de tontura
- palpitações cardíacas
- língua inflamada ou dolorida
- mãos e pés frios
Como testar anemia genética
O teste de anemia geralmente começa com um hemograma completo e um exame microscópico dessas células. Isso mostra o número e o formato dos glóbulos vermelhos, além dos níveis de hemoglobina.
Se houver alguma alteração incomum nos níveis de qualquer um deles, mais testes são realizados. Testes genéticos específicos para anemia também podem ser realizados analisando células retiradas de:
- Sangue simples
- Saliva
- Testes moleculares da medula ossea
Uma análise do seu DNA coletado dessas células pode determinar se você carrega ou não uma característica genética específica que resulta em alterações nos glóbulos vermelhos. Por exemplo, um teste genético para anemia falciforme testaria uma variação específica da hemoglobina conhecida como hemoglobina S.
Formas de tratar anemia
O tratamento para anemia pode variar de modificações na dieta até cirurgia. O tratamento específico que você pode precisar depende do tipo de anemia com que você vive.
A anemia causada por deficiências de nutrientes é tratada corrigindo a deficiência. Isso normalmente é realizado tomando suplementos vitamínicos ou comendo mais alimentos ricos em nutrientes específicos, como vitamina B12 ou ferro.
Anemias genéticas envolvem cuidados com alimentação, infecções e uso de medicações. Por isso, se você tem uma anemia genética, é importante o acompanhamento com médico especialista, no caso, hematologista.